While the clamour to declare thalassemia as a disability grows around the country, year 2015 has brought realisation and reaffirmation to the long held perspective that we have at Sankalp India Foundation for thalassemia.
As part of the counselling and motivational exercise, also as part of the speeches and the writings in the books, often the thalassemia families are told that their child should be treated like a normal child.
Is this really true? Is it a fact that the child suffering from thalassemia could actually have a near normal life? Or is it some kind of a motivational statement which seeks to overlook the reality of the situation?
 We have always believed that it is possible to restore the normalcy of life of a child suffering from thalassemia. We have always held to the notion that suffering from thalassemia does not make a child someone who cannot dream of a bright future. However, mismanagement of thalassemia is what causes pain and suffering.
Recent medical advances have ensured that the scenario that lies ahead for each person born with thalassemia has changed completely. There has been remarkable progress with blood safety and availability. The availability of oral chelators in the market has ensured that the need for infusion of drugs is limited now. Diagnostic facilities, prevention of co-morbidities (additional medical problems) and systematic disease management has been made possible by the advances in medical science, technology and medical education. In short, if a child is born with thalassemia today, he or she could have a normal education, could marry, have children, have a regular job, retire and die of natural reasons. Thalassemia is no longer a severely limiting disease nor is it a terminal illness.
Let us all say together and in clear loud terms – THALASSEMIA IS NOT A DISEASE! It is a DISORDER!
In this perspective, instead of worrying about how to justify thalassemia as disability and then seek some unrelated benefits and concessions, probably the debate should be more around how to ensure that the possibility of a near normal life is realised for each and every child. The need of the hour if strengthening of thalassemia management aggressively. We need to make sure that the advances made by the torchbearers in medicine and technology percolate down to the last child born with thalassemia. If this is done, suffering would be minimised, damage would be contained and normalcy restored.
The “utopian dream” as some would like to think of it, is not that difficult and unachievable after all. Last 4 years of working in the field of thalassemia has given us 3 key learning which would we like to share.
Delivery of Care should be Focused and Patient Centric
Chronic disease management, like that required for thalassemia, requires a comprehensive and robust approach to deliver care and management to the patient. The patient needs to be kept at the center stage and all the elements of care and management need to be brought together in one place rather than the patient having to seek them from place to place. Any person who walks into a healthcare facility for blood transfusion must get it in a reasonable amount of time. What’s more is that the doctor’s evaluations, lab tests, consultations, counselling, access to drugs, preventive elements of thalassemia and the option of cure, all of these individual elements should be planned such that they are delivered within that one single visit. A patient must spend the minimum possible time for things associated with disease management.
Is this possible? We have made this possible in last few years by organising the care for thalassemia in day care centers which specialise in the management of blood disorders. These centers are designed and developed to ensure that each element of care is optimised both for the care giver and receiver for quality, affordability and reliability. We have demonstrated over the years how it is possible to deliver state of the art thalassemia care and management in-line with the internationally acceptable guidelines in a sustained manner even at one fifth the cost (or less) at which it is available otherwise. We have extensively used technology to foster quality and reliability. Our centers are powered by the world’s first ever software package which enables end to end management of thalassemia. The procedures, data management and work planning has been simplified and streamlined ensuring that the facilities are able to optimally use the resources, be it the physical resources or the manpower.
The setting up of a thalassemia day care centre at Indira Gandhi Institute of Child Health ensured that the number of children receiving treatment increased from 65 to 296. The pre-transfusion haemoglobin increased from 6 gm/dl to 8.5 gm/dl and the access to systematic chelation increased from 20 kids to about 231 kids in a matter of 3 years. Within 7 hours the patients get transfusion, chelation, lab tests and meet with the doctors.
Damage is reversible
If a young child who get’s diagnosed with thalassemia today, comes to a particular healthcare setup for treatment, and still lands up with high ferritin, large liver, large spleen and other complications associated with thalassemia, clearly it is complete failure of the basic medical treatment approach and delivery. However, the question that has been haunting the fraternity associated with thalassemia is what to do for the individuals who have not received proper care and management for years.
Sankalp India Foundation received most of the children who had never received proper chelation. The persistently low haemoglobin levels had taken the toll on the children and the overall assessment at the time of joining the day cares was not good. However, the good news is that it is clear that it is indeed possible to reverse a large number of complications which are seen by persistent effort and modern therapy. Sustained hyper transfusion has huge positive impact. Aggressive use of chelators with strict monitoring reverses the worrisome trends. And highlight of our experience has been the positive impact of hydroxyurea. We have used hydroxyurea not just to trigger the production of fetal haemoglobin in the thalassemia intermedias but also to correct the hepatomegaly, splenomagaly and also to assist ferritin control in thalassemia majors.
Unfortunately this cannot be said for each child. We still struggle to contain the damage if not reverse it in certain situations, but largely the experience of reversing the damage has been very positive.
We have always believed that it is possible to restore the normalcy of life of a child suffering from thalassemia. We have always held to the notion that suffering from thalassemia does not make a child someone who cannot dream of a bright future. However, mismanagement of thalassemia is what causes pain and suffering.
Recent medical advances have ensured that the scenario that lies ahead for each person born with thalassemia has changed completely. There has been remarkable progress with blood safety and availability. The availability of oral chelators in the market has ensured that the need for infusion of drugs is limited now. Diagnostic facilities, prevention of co-morbidities (additional medical problems) and systematic disease management has been made possible by the advances in medical science, technology and medical education. In short, if a child is born with thalassemia today, he or she could have a normal education, could marry, have children, have a regular job, retire and die of natural reasons. Thalassemia is no longer a severely limiting disease nor is it a terminal illness.
Let us all say together and in clear loud terms – THALASSEMIA IS NOT A DISEASE! It is a DISORDER!
In this perspective, instead of worrying about how to justify thalassemia as disability and then seek some unrelated benefits and concessions, probably the debate should be more around how to ensure that the possibility of a near normal life is realised for each and every child. The need of the hour if strengthening of thalassemia management aggressively. We need to make sure that the advances made by the torchbearers in medicine and technology percolate down to the last child born with thalassemia. If this is done, suffering would be minimised, damage would be contained and normalcy restored.
The “utopian dream” as some would like to think of it, is not that difficult and unachievable after all. Last 4 years of working in the field of thalassemia has given us 3 key learning which would we like to share.
Delivery of Care should be Focused and Patient Centric
Chronic disease management, like that required for thalassemia, requires a comprehensive and robust approach to deliver care and management to the patient. The patient needs to be kept at the center stage and all the elements of care and management need to be brought together in one place rather than the patient having to seek them from place to place. Any person who walks into a healthcare facility for blood transfusion must get it in a reasonable amount of time. What’s more is that the doctor’s evaluations, lab tests, consultations, counselling, access to drugs, preventive elements of thalassemia and the option of cure, all of these individual elements should be planned such that they are delivered within that one single visit. A patient must spend the minimum possible time for things associated with disease management.
Is this possible? We have made this possible in last few years by organising the care for thalassemia in day care centers which specialise in the management of blood disorders. These centers are designed and developed to ensure that each element of care is optimised both for the care giver and receiver for quality, affordability and reliability. We have demonstrated over the years how it is possible to deliver state of the art thalassemia care and management in-line with the internationally acceptable guidelines in a sustained manner even at one fifth the cost (or less) at which it is available otherwise. We have extensively used technology to foster quality and reliability. Our centers are powered by the world’s first ever software package which enables end to end management of thalassemia. The procedures, data management and work planning has been simplified and streamlined ensuring that the facilities are able to optimally use the resources, be it the physical resources or the manpower.
The setting up of a thalassemia day care centre at Indira Gandhi Institute of Child Health ensured that the number of children receiving treatment increased from 65 to 296. The pre-transfusion haemoglobin increased from 6 gm/dl to 8.5 gm/dl and the access to systematic chelation increased from 20 kids to about 231 kids in a matter of 3 years. Within 7 hours the patients get transfusion, chelation, lab tests and meet with the doctors.
Damage is reversible
If a young child who get’s diagnosed with thalassemia today, comes to a particular healthcare setup for treatment, and still lands up with high ferritin, large liver, large spleen and other complications associated with thalassemia, clearly it is complete failure of the basic medical treatment approach and delivery. However, the question that has been haunting the fraternity associated with thalassemia is what to do for the individuals who have not received proper care and management for years.
Sankalp India Foundation received most of the children who had never received proper chelation. The persistently low haemoglobin levels had taken the toll on the children and the overall assessment at the time of joining the day cares was not good. However, the good news is that it is clear that it is indeed possible to reverse a large number of complications which are seen by persistent effort and modern therapy. Sustained hyper transfusion has huge positive impact. Aggressive use of chelators with strict monitoring reverses the worrisome trends. And highlight of our experience has been the positive impact of hydroxyurea. We have used hydroxyurea not just to trigger the production of fetal haemoglobin in the thalassemia intermedias but also to correct the hepatomegaly, splenomagaly and also to assist ferritin control in thalassemia majors.
Unfortunately this cannot be said for each child. We still struggle to contain the damage if not reverse it in certain situations, but largely the experience of reversing the damage has been very positive.
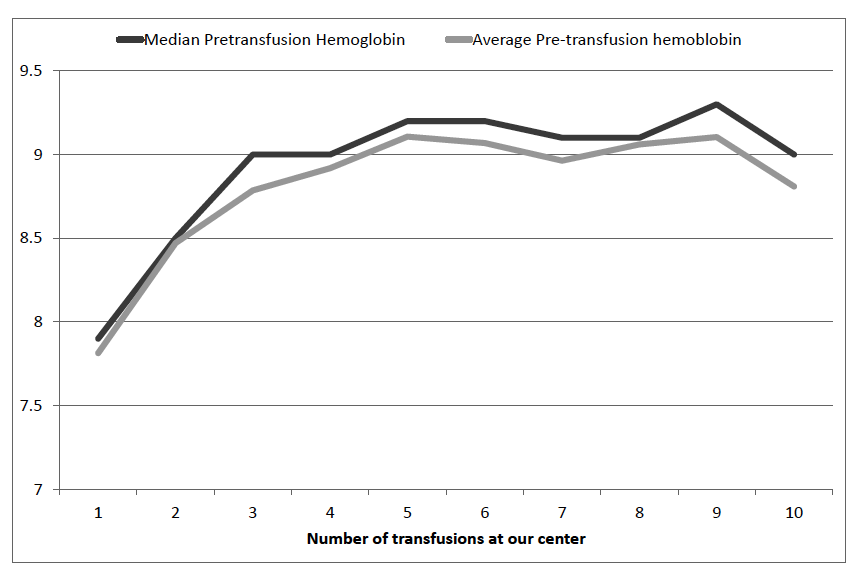 Chart showing the correction in pre-transfusion hemoglobin at Samraksha - thalassemia day care centre across first ten transfusions.
The option of cure is real
“Young TM patients with an available HLA identical sibling should be offered HSCT as soon as possible before development of iron overload and iron-related tissue damage.”
- (Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 2014;99(5):811–20.)
Bone marrow transplantation has been an established and reliable cure from thalassemia in the western world. The cost, limited access and poor outcomes in Indian setting have ensured that to continue to remain out of reach for most individuals suffering from thalassemia. Leave alone the transplant, the first step of HLA typing to determine whether a transplant is possible or not is also considered out of reach for most children. In the last 3 years we have participated and experienced in changing all of this. At our thalassemia centers, every child who has a healthy sibling is being offered HLA typing. Those who get a match are being offered transplantations. For those who did proceed with the transplantations, the outcome so far has been the best possible (100% cure). This is a real leap as far as hope and access to cure is concerned.
Chart showing the correction in pre-transfusion hemoglobin at Samraksha - thalassemia day care centre across first ten transfusions.
The option of cure is real
“Young TM patients with an available HLA identical sibling should be offered HSCT as soon as possible before development of iron overload and iron-related tissue damage.”
- (Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 2014;99(5):811–20.)
Bone marrow transplantation has been an established and reliable cure from thalassemia in the western world. The cost, limited access and poor outcomes in Indian setting have ensured that to continue to remain out of reach for most individuals suffering from thalassemia. Leave alone the transplant, the first step of HLA typing to determine whether a transplant is possible or not is also considered out of reach for most children. In the last 3 years we have participated and experienced in changing all of this. At our thalassemia centers, every child who has a healthy sibling is being offered HLA typing. Those who get a match are being offered transplantations. For those who did proceed with the transplantations, the outcome so far has been the best possible (100% cure). This is a real leap as far as hope and access to cure is concerned.
 "There is no doubt that people with thalassemia have a growing number of options for well being and cure, we just need to work together to make them available."
Dr. Lawrence Faulkner
Program Director, Sankalp-People Tree Hospitals Bone Marrow Transplant Unit, Bangalore, India
Transplantation for thalassemia is becoming safer and more reliable by the day. Again, with the blend of medicine, technology and the right team, it is indeed possible to achieve world class outcome (> 95% overall survival and >90% cure) which compares well with the outcomes of the available supportive care.
So far we have done 13 transplants for children suffering from thalassemia. All of them are doing well and are transfusion free.
Even for thalassemiacs who are currently not the best candidates for transplantation because of their age or medical condition, the progress in the last few years has been very promising. Improved, low-intensity conditioning regimens (therapy used for transplants) have ensured that the age at which the transplants can be safely offered is increasing. For those who do not have a sibling donor, the beam of hope comes from the positive experiences from the haplo-identical transplantations from some centers (who have managed to get it right so far). Once haplo-identical transplantation is available with comparable outcomes to matched sibling donors, potentially, each patient will have an option for complete cure. From what we understand, this situation is not too far into the future.
"There is no doubt that people with thalassemia have a growing number of options for well being and cure, we just need to work together to make them available."
Dr. Lawrence Faulkner
Program Director, Sankalp-People Tree Hospitals Bone Marrow Transplant Unit, Bangalore, India
Transplantation for thalassemia is becoming safer and more reliable by the day. Again, with the blend of medicine, technology and the right team, it is indeed possible to achieve world class outcome (> 95% overall survival and >90% cure) which compares well with the outcomes of the available supportive care.
So far we have done 13 transplants for children suffering from thalassemia. All of them are doing well and are transfusion free.
Even for thalassemiacs who are currently not the best candidates for transplantation because of their age or medical condition, the progress in the last few years has been very promising. Improved, low-intensity conditioning regimens (therapy used for transplants) have ensured that the age at which the transplants can be safely offered is increasing. For those who do not have a sibling donor, the beam of hope comes from the positive experiences from the haplo-identical transplantations from some centers (who have managed to get it right so far). Once haplo-identical transplantation is available with comparable outcomes to matched sibling donors, potentially, each patient will have an option for complete cure. From what we understand, this situation is not too far into the future.
 In summary, our experience has been very promising for the thalassemiacs. We need a mindset which focussed on getting the necessary elements of care, management and cure in right shape and delivering to every child. Thalassemia today, is manageable and curable. We hate to call it a disability because we believe in restoring normalcy!
Of-course a lot of work remains. Of-course the road is long. However, today the question is that of direction. A direction which empowers individuals, strengthens their ability to pursue their dreams, actually makes them near normal , not just aim at making them feel normal is the direction we choose.
In summary, our experience has been very promising for the thalassemiacs. We need a mindset which focussed on getting the necessary elements of care, management and cure in right shape and delivering to every child. Thalassemia today, is manageable and curable. We hate to call it a disability because we believe in restoring normalcy!
Of-course a lot of work remains. Of-course the road is long. However, today the question is that of direction. A direction which empowers individuals, strengthens their ability to pursue their dreams, actually makes them near normal , not just aim at making them feel normal is the direction we choose.
We have always believed that it is possible to restore the normalcy of life of a child suffering from thalassemia. We have always held to the notion that suffering from thalassemia does not make a child someone who cannot dream of a bright future. However, mismanagement of thalassemia is what causes pain and suffering.
Recent medical advances have ensured that the scenario that lies ahead for each person born with thalassemia has changed completely. There has been remarkable progress with blood safety and availability. The availability of oral chelators in the market has ensured that the need for infusion of drugs is limited now. Diagnostic facilities, prevention of co-morbidities (additional medical problems) and systematic disease management has been made possible by the advances in medical science, technology and medical education. In short, if a child is born with thalassemia today, he or she could have a normal education, could marry, have children, have a regular job, retire and die of natural reasons. Thalassemia is no longer a severely limiting disease nor is it a terminal illness.
Let us all say together and in clear loud terms – THALASSEMIA IS NOT A DISEASE! It is a DISORDER!
In this perspective, instead of worrying about how to justify thalassemia as disability and then seek some unrelated benefits and concessions, probably the debate should be more around how to ensure that the possibility of a near normal life is realised for each and every child. The need of the hour if strengthening of thalassemia management aggressively. We need to make sure that the advances made by the torchbearers in medicine and technology percolate down to the last child born with thalassemia. If this is done, suffering would be minimised, damage would be contained and normalcy restored.
The “utopian dream” as some would like to think of it, is not that difficult and unachievable after all. Last 4 years of working in the field of thalassemia has given us 3 key learning which would we like to share.
Delivery of Care should be Focused and Patient Centric
Chronic disease management, like that required for thalassemia, requires a comprehensive and robust approach to deliver care and management to the patient. The patient needs to be kept at the center stage and all the elements of care and management need to be brought together in one place rather than the patient having to seek them from place to place. Any person who walks into a healthcare facility for blood transfusion must get it in a reasonable amount of time. What’s more is that the doctor’s evaluations, lab tests, consultations, counselling, access to drugs, preventive elements of thalassemia and the option of cure, all of these individual elements should be planned such that they are delivered within that one single visit. A patient must spend the minimum possible time for things associated with disease management.
Is this possible? We have made this possible in last few years by organising the care for thalassemia in day care centers which specialise in the management of blood disorders. These centers are designed and developed to ensure that each element of care is optimised both for the care giver and receiver for quality, affordability and reliability. We have demonstrated over the years how it is possible to deliver state of the art thalassemia care and management in-line with the internationally acceptable guidelines in a sustained manner even at one fifth the cost (or less) at which it is available otherwise. We have extensively used technology to foster quality and reliability. Our centers are powered by the world’s first ever software package which enables end to end management of thalassemia. The procedures, data management and work planning has been simplified and streamlined ensuring that the facilities are able to optimally use the resources, be it the physical resources or the manpower.
The setting up of a thalassemia day care centre at Indira Gandhi Institute of Child Health ensured that the number of children receiving treatment increased from 65 to 296. The pre-transfusion haemoglobin increased from 6 gm/dl to 8.5 gm/dl and the access to systematic chelation increased from 20 kids to about 231 kids in a matter of 3 years. Within 7 hours the patients get transfusion, chelation, lab tests and meet with the doctors.
Damage is reversible
If a young child who get’s diagnosed with thalassemia today, comes to a particular healthcare setup for treatment, and still lands up with high ferritin, large liver, large spleen and other complications associated with thalassemia, clearly it is complete failure of the basic medical treatment approach and delivery. However, the question that has been haunting the fraternity associated with thalassemia is what to do for the individuals who have not received proper care and management for years.
Sankalp India Foundation received most of the children who had never received proper chelation. The persistently low haemoglobin levels had taken the toll on the children and the overall assessment at the time of joining the day cares was not good. However, the good news is that it is clear that it is indeed possible to reverse a large number of complications which are seen by persistent effort and modern therapy. Sustained hyper transfusion has huge positive impact. Aggressive use of chelators with strict monitoring reverses the worrisome trends. And highlight of our experience has been the positive impact of hydroxyurea. We have used hydroxyurea not just to trigger the production of fetal haemoglobin in the thalassemia intermedias but also to correct the hepatomegaly, splenomagaly and also to assist ferritin control in thalassemia majors.
Unfortunately this cannot be said for each child. We still struggle to contain the damage if not reverse it in certain situations, but largely the experience of reversing the damage has been very positive.
"There is no doubt that people with thalassemia have a growing number of options for well being and cure, we just need to work together to make them available."
Dr. Lawrence Faulkner
Program Director, Sankalp-People Tree Hospitals Bone Marrow Transplant Unit, Bangalore, India
Transplantation for thalassemia is becoming safer and more reliable by the day. Again, with the blend of medicine, technology and the right team, it is indeed possible to achieve world class outcome (> 95% overall survival and >90% cure) which compares well with the outcomes of the available supportive care.
So far we have done 13 transplants for children suffering from thalassemia. All of them are doing well and are transfusion free.
Even for thalassemiacs who are currently not the best candidates for transplantation because of their age or medical condition, the progress in the last few years has been very promising. Improved, low-intensity conditioning regimens (therapy used for transplants) have ensured that the age at which the transplants can be safely offered is increasing. For those who do not have a sibling donor, the beam of hope comes from the positive experiences from the haplo-identical transplantations from some centers (who have managed to get it right so far). Once haplo-identical transplantation is available with comparable outcomes to matched sibling donors, potentially, each patient will have an option for complete cure. From what we understand, this situation is not too far into the future.
In summary, our experience has been very promising for the thalassemiacs. We need a mindset which focussed on getting the necessary elements of care, management and cure in right shape and delivering to every child. Thalassemia today, is manageable and curable. We hate to call it a disability because we believe in restoring normalcy!
Of-course a lot of work remains. Of-course the road is long. However, today the question is that of direction. A direction which empowers individuals, strengthens their ability to pursue their dreams, actually makes them near normal , not just aim at making them feel normal is the direction we choose.
Patrika Edition
Patrika Section
Disqus Comment